I need to update my previous post on how to install gromacs (gmx). There is one more package you need to install with cygwin: tcsh
Apparently this is required to run csh scripts, which you need to do. If you have already installed cygwin, you can add the package to it by running the setup again and selecting the package. If you want to know what packages you have installed, you can use: cygcheck -cd
Now, there is a built in demonstration script that comes with gmx. It is located in:
[your path]/gromacs/tutor/gmxdemo (as you are about to see my path is rather redicioulous)
1. fire up cygwin
2 change your directory to gmxdemo:
cd c:cygwin/usr/local/gromacs/share/gromacs/tutor/gmxdemo
3. after you have the tcsh package installed, all you have to do is type:
./demo
This should start the demo and you'll get:
-----------------------------------------------------------------
-----------------------------------------------------------------
Welcome to the GROMACS demo.
This is a script that takes about 10 min to run.
In this demo we will demonstrate how to simulate Molecular
Dynamics (MD) using the GROMACS software package.
The demo will perform a complete molecular dynamics (MD) simulation
of a small peptide in water. The only input file we need to do this
is a pdb file of a small peptide.
If you have any problems or remarks with respect to this demonstration,
please mail to: gromacs@gromacs.org , or check the resources on
our website http://www.gromacs.org .
-----------------------------------------------------------------
-----------------------------------------------------------------
Press
Then just follow the instructions, press enter when it says, pull on your beard every couple of seconds, hoping it doesn't choke again.
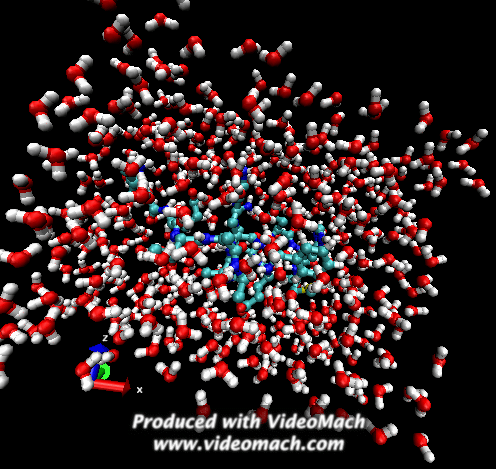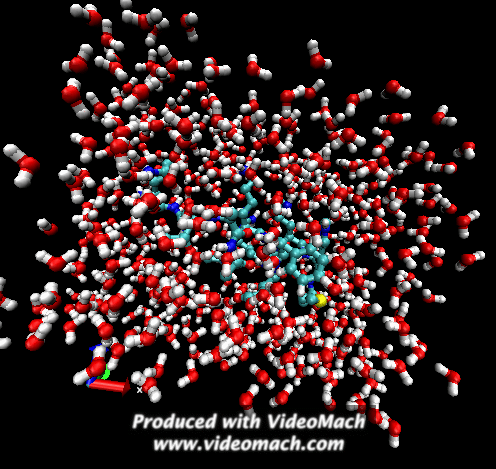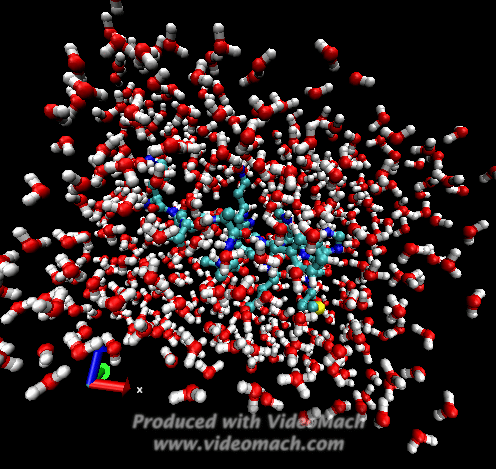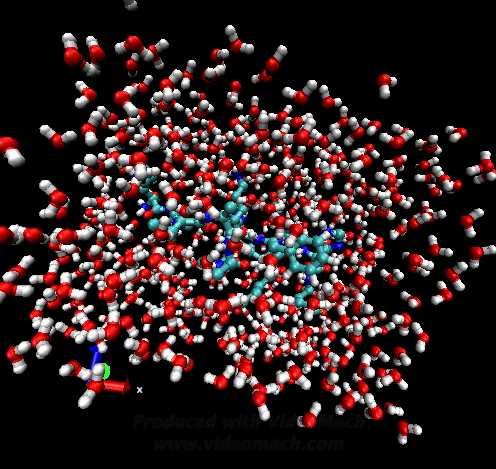
When the demo is finished, it'll ask you to start another program to view the results. I didn't. I used VMD to look at and make the above animation. And as you can tell there was a little help from videomach. There are lots of other programs to do this, just trying some thing new.
Once you have VMD installed:
1. From the main console: file>new molecule
2. load cpeptide_after_md.gro
3. in the main console, select cpeptide_after_md and file>load data into molecule
4. load cpeptide_md.trr this is the gmx trajectory file
5. use the play bar at the bottom of main to view the results.
MTF

No comments:
Post a Comment